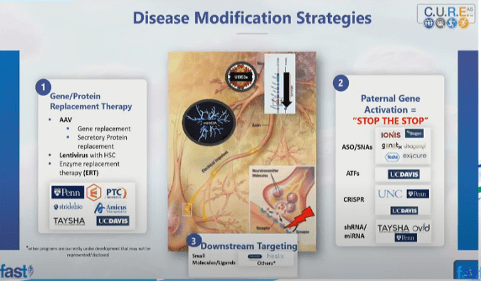

1. Parliamo della terapia genica, quindi dell’utilizzo dell’AAV. L’AAV è un virus adeno-associato, non patogeno, in cui è stato inserito il gene UBE3A, che viene trasportato nel sistema nervoso centrale o nel cervello, dove verrà espresso e, quindi, produrrà la proteina mancante. La terapia genica è stata descritta per la prima volta dall’Università del Sud della Florida dal Dott. Weeber e dal Dott. Nash (anno 2011). Anche UPenn ha riportato dati su modelli murini di AS con terapia AAV, così come l’Università del Nord Carolina in collaborazione con Taysha Pharmaceutical.
Anderson ha da poco riportato (2021) il programma di terapia genica con le cellule staminali ematopoietiche che hanno avuto una notevole risposta nel modello animale. Ci sono quattro programmi aggiuntivi attivamente perseguiti da diverse ditte farmaceutiche o partner accademici.

Questo è un esempio di risposta dei modelli murini alla terapia genica. La slide è tratta da un paper pubblicato da poco (ottobre 2021) da Matthew Judson e Ben Philpot in cui ci sono due modelli murini, uno senza (WT) e l’altro con la sindrome di Angelman. Si nota come il topo WT può creare un nido con la carta, a differenza del topo con la SA che non può costruire nessun nido. Tuttavia il modello murino con la sindrome di Angelman trattato con la terapia genica (quindi con l’AAV) acquisisce la capacità di costruire un nido. È piuttosto impressionante come si passi dall’incompleta capacità di formare un nido con la carta ad esserne invece in grado dopo il trattamento.
Vi è anche un esempio di terapia genica con le cellule staminali ematopoietiche del laboratorio di Anderson e del laboratorio di Silverman. I topi con la sindrome hanno difficoltà a coordinarsi e stare al passo su un tapis roulant, quindi colpiscono il paraurti senza sosta. Il topo dopo la terapia genica è totalmente differente, riacquista la capacità di coordinazione caratteristica di un topo sano.

Ora parliamo un po’ della terapia enzimatica sostitutiva. Vi è un farmaco approvato da BioMarin, che viene messo proprio a livello del cuoio capelluto e la proteina UBE3A passa direttamente nel cervello, dove potrebbe funzionare sia a livello dei neuroni (uptalke cellulare) che a livello sinaptico (tra i neuroni). Queste osservazioni però sono ancora qualcosa di sperimentale perché potrebbe trattarsi di una terapia che non dura per tutta la vita ma potrebbe richiedere diverse somministrazioni.

2. La seconda strategia vista è quella di attivare la copia paterna. La prima persona a riconoscere in questa strategia una valida opzione è stato il Dott. Ben Philpot dell’Università del Nord Carolina Chapel Hill, che nel 2011 ha pubblicato l’uso del Topotecan, un inibitore della topoisomerasi in grado di abbattere il trascritto antisenso e attivare la copia paterna del gene. Ma non è stato possibile utilizzare l’inibitore clinicamente in un modello murino perché era molto tossico, quindi non sapevamo come sarebbero stati i risultati.
Il Dott. Art Beaudet è stato il primo a pubblicare l’uso degli oligonucleotidi antisenso (ASO) nel 2015 in un modello murino di sindrome di Angelman, ed è stato in grado di dimostrare la reversione del fenotipo in alcuni topi con la sindrome. Infine, il Dott. Dindot nel 2021 ha pubblicato recentemente un ASO per la sindrome di Angelman.
Il Dott. David Segal ha pubblicato l’uso di fattori di trascrizione artificiali nell’anno 2016, mentre il Dott. Butler e il Dott. Chamberlain stanno entrambi lavorando su un approccio a shRNA per la sindrome di Angelman. Infine, il Dott. Wilson e il suo team stanno lavorando su un approccio con i miRNA come terapia per l’AS.
Si può concludere dicendo che abbiamo tantissimi approcci possibili per attivare la copia paterna.

Nelle immagini potete vedere tutti i progetti. Quello del Dott. Art Beaudet qui sopra, che ha fatto uso degli ASOs come sistema di attivazione della copia paterna, ha ottenuto una risposta al deficit cognitivo nel modello murino in seguito all’iniezione diretta nel fluido cerebrale.
In pratica, in questo momento abbiamo due studi clinici sugli esseri umani in corso, condotti da Genetix in collaborazione con Ultragenyx e da Roche in collaborazione con Genentech negli Stati Uniti e in Europa.

Vediamo poi uno studio di attivazione del gene paterno mediante l’uso del fattore di trascrizione artificiale. Questo sistema è efficace nel determinare l’attivazione della copia paterna del gene UBE3A, ma è meno specifico rispetto agli ASOs, motivo per cui la comunità scientifica sta investendo di meno su questo approccio ed è più indietro come terapia rispetto agli ASOs stessi.
 Proseguiamo poi con il lavoro del Dott. Mark Zilka dell’Università del Nord Carolina che si basa sul sistema di CRISPR. Questo sistema ha dimostrato essere in grado di ottenere un recupero del fenotipo nel modello murino AS se somministrato precocemente, nello specifico durante lo sviluppo, facendo recuperare la maggior parte dei tratti fenotipici dell’Angelman.
Proseguiamo poi con il lavoro del Dott. Mark Zilka dell’Università del Nord Carolina che si basa sul sistema di CRISPR. Questo sistema ha dimostrato essere in grado di ottenere un recupero del fenotipo nel modello murino AS se somministrato precocemente, nello specifico durante lo sviluppo, facendo recuperare la maggior parte dei tratti fenotipici dell’Angelman.

3. Infine, non possiamo dimenticare i bersagli a valle perché potrebbero dimostrarsi abbastanza impattanti. Pensiamo al Gaboxadol, che era il farmaco in sperimentazione da parte dell’Ovid Therapeutics, ma che purtroppo non ha superato la fase 3 di sperimentazione clinica. Ci sono poi integratori chetonici con cui hanno condotto delle sperimetazioni cliniche. Hanno poi utilizzato ligandi o agonisti di IGF-1 e IGF-2 in NYU e ora le aziende farmaceutiche stanno lavorando su un analogo di IGF-1.

Questo è il trial NNZ-2591 che si spera andrà in sperimentazione clinica in brevissimo tempo in Australia. Quello che sono stati in grado di vedere è un enorme recupero delle attività del topo con la sindrome di Angelman. Questo farmaco non è altro che una pillola o un liquido che può essere somministrato e indurre una corretta regolazione tra le sinapsi neuronali in modo che le cellule neuronali possano parlare tra di loro in modo migliore.

Guardando questa diapositiva potete confrontare il colore rosso (no recupero del fenotipo) con il verde (si recupero del fenotipo). Quello che vorrei a questo punto è che apprezzaste tutti gli studi di cui ho appena parlato. Questi sono tutti i test dove c’è stato il recupero del fenotipo murino. Questo è stato un decennio incredibile perché abbiamo curato i sintomi in diversi modi.
Il lavoro presente nell’ultima colonna mostra il recupero completo di tutte le disfunzioni in un modello murino adulto, ma ci sono altri casi in cui si possono recuperare alcune funzioni mentre altre no. Questi dati contraddittori sono dovuti al fatto che il modello animale utilizzato non è perfetto e, quindi, è vincolante la finestra di età in cui si va ad agire con il trattamento. Per questo motivo, dobbiamo uscire rapidamente dal modello murino e iniziare a capire cosa possiamo fare sugli esseri umani.

Come ci prepariamo per i futuri trial? Mi concentrerò principalmente sui trial dove ora si stanno concentrando i vostri finanziamenti.
Dobbiamo creare un modello animale e delle linee cellulari per i test preclinici. Finora, infatti, abbiamo finanziato la creazione del modello di ratto dell’AS, il modello con la delezione, per testare davvero il comportamento e capire cosa cambia nella sindrome di Angelman. Questo è stato supportato dalla UC Davis e USF. Il modello del ratto è sicuramente un modello animale migliore di quello murino in diverse patologie.
Abbiamo creato anche un modello di maiale per la sindrome di Angelman e un modello murino con una grande delezione (5Mb), che include non solo UBE3A ma anche i geni limitrofi (circa 10 geni). Quest’ultimo modello è stato prodotto per capire il significato clinico di questi geni e che impatto hanno quando deleti. E, per completezza, abbiamo un altro modello murino in cui mancano i 10 geni ma non UBE3A. Infine, abbiamo creato linee cellulari iPSC umane grazie alle vostre donazioni che ora vengono distribuite in tutto il mondo a molte aziende farmaceutiche e partner accademici e che in molti laboratori possono essere utilizzate come test per diversi farmaci.
Ora stiamo finanziando presso la NYU anche un altro modello di linea cellulare iPSC per capire l’impatto dei geni e/o dell’RNA al di fuori di UBE3A. In ultimo, si stanno creando anche organoidi al fine di testare la comunicazione tra i neuroni di ogni genotipo, senza dover necessariamente ricorre all’utilizzo di modelli animali. Il Dott. King sta anche facendo linee cellulari reporter in modo da essere in grado di osservare l’attivazione del gene paterno (rosso o verde brillante) da parte del farmaco; questo è sicuramente un metodo veloce per noi per fare screening ad alto rendimento per i farmaci che si vogliono testare.

Solo nel 2020 abbiamo finanziato la creazione del programma per la sindrome di Angelman presso l’UC Davis, che ha lo scopo di fornire un servizio a supporto dei test farmacologici preclinici per molte aziende farmaceutiche e partner accademici. Questa è un’istituzione che conosce il modello animale incredibilmente bene. Ciò che fanno è prendere un farmaco da chiunque lo produca e testarlo nei loro modelli animali (es. topo, ratto, linee cellulari), in modo tale da fare in seguito un resoconto all’azienda se il determinato farmaco funziona o meno, se permette di recuperare un determinato fenotipo piuttosto che un altro, ecc..
In un anno di finanziamenti, cinque aziende farmaceutiche hanno sostenuto il loro sviluppo di farmaci con l’idea che esiste questa infrastruttura che permette, con le conoscenze di biologia molecolare e dei modelli animali, di fare screening di farmaci ad ampio spettro, ottimizzando tempo e denaro.

Pensiamo al punto in cui eravamo e dove siamo ora. Abbiamo iniziato con la ricerca di base, siamo passati alla scoperta di farmaci, arrivando al preclinico e poi, con un candidato, abbiamo proseguito con gli studi di tossicologia, chiamati IND Enabling Studies, serviti per approvare cioè che può essere o meno somministrato all’uomo. Una volta approvato, il farmaco può passare agli studi di trial clinico: la fase 1 è la valutazione della sicurezza e tollerabilità, la fase 2 è la valutazione dell’attività terapeutica del potenziale farmaco, cioè la sua capacità di produrre sull’organismo umano gli effetti curativi desiderati, e la fase 3 è la determinazione dell’efficacia e nuovamente della sicurezza del farmaco.

Quindi, lo sviluppo dei farmaci avviene in questo modo. Si passa da uno screening di circa 15.000 farmaci, fino a circa 5.000 farmaci che passano poi ai trial clinici. Appena una media di 10 farmaci riescono a passare alla sperimentazione clinica e solo 1 ottiene l’approvazione. Dobbiamo finanziare ogni passaggio di questo lungo processo e fare in modo che non solo un farmaco, ma più di uno, venga approvato. FAST è qui per supportare la ricerca sperimentale che possa condurre all’approvazione di farmaci per ciascun genotipo della sindrome di Angelman e, grazie al FAST FIRE TEAM (2013), siamo riusciti ad avanzare molto nella ricerca di base, nella scoperta di nuovi farmaci e nel trovare candidati che riuscivano a passare questi studi. Poi c’è stata la FAST infrastructure Program (2020) che ha accelerato il passaggio da uno studio preclinico a un candidato più vicino all’uomo. Infine ci sono gli IND enabling studies ultimo step prima della sperimentazione clinica, che è sempre stato il nostro obiettivo finale.

Qui sopra quello che eravamo 6 anni fa, quando abbiamo avuto un AAV, un ASO, un fattore di trascrizione artificiale e due farmaci per agire sui pathway a valle, eravamo nella finestra preclinica.

Due anni fa ci siamo mossi verso gli studi clinici, soprattutto con i downstream target. Gli studi in questa finestra temporale sono progrediti molto velocemente.

Ecco oggi come appare lo scenario. È incredibile dove siamo arrivati in un tempo così breve. Con i trial clinici, purtroppo, arrivano anche alcuni fallimenti. Di certo non dobbiamo scoraggiarci e, infatti, quello su cui mi voglio soffermare sono i puntini neri, che rappresentano ognuno uno studio terapeutico per la sindrome di Angelman. Questi risultati sono stati accelerati grazie a voi che avete investito tempo e denaro e grazie ai nostri scienziati per il loro enorme contributo nella ricerca traslazionale di questa sindrome
Grazie”.
Allyson Berent
Riassunto di Ornella Rondinone (Università di Milano)
